




INTRODUCTION
Synovial sarcomas (SS) account for almost 10% of all soft tissue sarcomas and typically arise in the para-articular regions, but actually has been shown to have no biologic or pathologic relationship to synovium. A subtype, primary pleural synovial sarcoma (PPSS),which is thought to arise from the pleura is idiopathic and extremely rare. PPSS is a locally aggressive tumor, seen most frequently in patients over age 40 years, compared to other SS which more commonly occur in adolescents and young adults.1 The diagnosis of PPSS is made after excluding metastasis from an extra-thoracic site.
We report the case of a patient who presented with a mass bridging the upper retroperitoneum and lower posterior mediastinum and involving the adjacent pleura. We discuss the surgical approach and management of this unusual case within the context of the published literature. Written informed consent was obtained from the patient for publication of this case report and accompanying images.
CASE SUMMARY
A 55-year-old female patient had repeated visits to an outside hospital over six months with complaints of lower back pain and abdominal fullness. She denied dysphagia and weight loss. She had no other significant medical or surgical history. Computed tomography (CT) demonstrated a possible distal esophageal tumor. She was referred to our hospital for consideration of surgical resection.
On physical examination, the patient was in no acute distress, had normal vital signs and no appreciable abdominal mass or tenderness. Hematologic profile, electrolytes, liver, and renal function tests were normal. Contrast CT of the chest and upper abdomen revealed a 7 cm x 5.5 cm x 4.5 cm well circumscribed lower mediastinal and upper retroperitoneal soft tissue mass with internal calcifications either adjacent to or arising from the distal esophagus (Figures 1a and 1b). Upper gastrointestinal endoscopy (EGD) was unremarkable.


Based on the clinical presentation, relatively indolent course and CT findings, the presumptive preoperative diagnoses were paraganglioma or gastrointestinal stromal tumor (GIST). Following multidisciplinary discussions with our radiologists, percutaneous needle biopsy for diagnosis was not considered technically feasible. There is also no access currently to esophageal ultrasound guided biopsies at our hospital so it was decided to proceed directly to surgical exploration and resection.
Surgical Approach
After placing an epidural catheter, general anesthesia was induced and the patient was placed in the partial left lateral decubitus position (right side elevated approximately 30 degrees), a right thoracoabdominal incision was made. The right 9th intercostal space was entered, the costal margin divided, and the incision extended in the midline of the upper abdomen to the level of the umbilicus. The right lung was deflated and the right pleural space entered. The diaphragm was incised parallel and close to the costal margin to access the peritoneal cavity. Thus there was adequate exposure of the gastroesophageal junction, hepatic veins, inferior vena cava and inferior aspect of the pulmonary hilum.
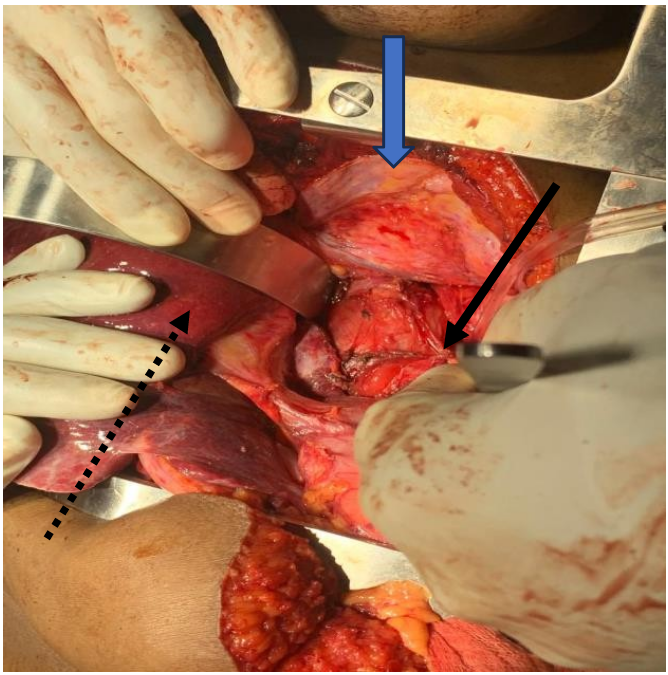
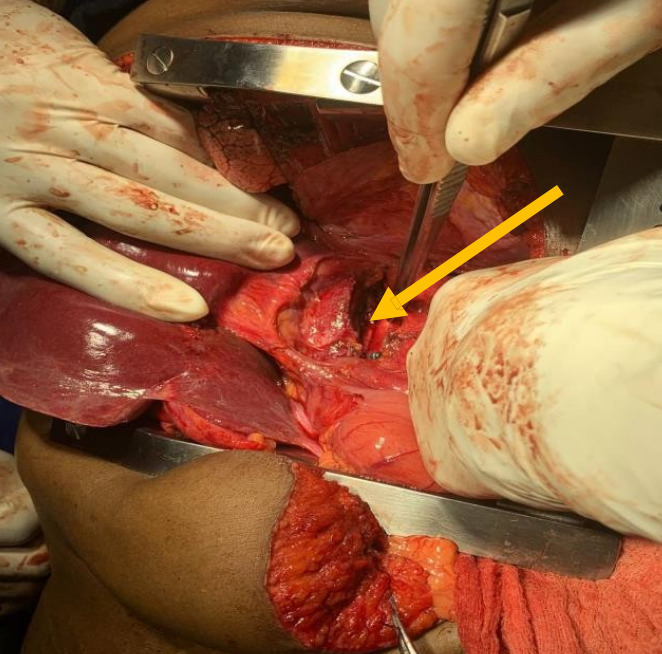
Intraoperative findings
The mass was well circumscribed with moderate peritumoral fibrosis, and was located in the posterior mediastinum and upper retroperitoneum with adhesion to the right crus of the diaphragm, and adhesion and leftward displacement of the distal thoracic and abdominal esophagus. It abutted the hepatic veins and inferior vena cava. There was no evidence of invasion of the esophagus, proximal stomach, liver, thoracic or lumbar vertebrae (Figures 2a and 2b). There were no intra-thoracic or peritoneal metastases.


The right pleura was widely opened, the inferior pulmonary ligament divided, the lung retracted superiorly, and the diaphragmatic incision extended to expose the intra- abdominal component of the mass. The lower thoracic esophagus above the tumor was encircled and retracted towards to the patient’s left to expose the mass which was gradually dissected circumferentially, freed from the crura, esophagus, spine, hepatic veins, inferior vena cava, and descending thoracic aorta, and completely removed. The incision was closed in the standard manner with interrupted non-absorbable horizontal mattress sutures to reapproximate the diaphragm, and closure in layers of the thoracic and abdominal components of the incision.
Pathology findings
Grossly, the tumor measured 7x6x3cm, appeared solid and encapsulated (Figure 3a). Histologic examination (by hematoxylin and eosin staining) showed a high-grade sarcoma consistent with biphasic synovial sarcoma. Microscopic evaluation showed solid proliferation of plump spindle cells with hyperchromatic nuclei and variable sized nests of glands lined by columnar cells, some glands containing eosinophilic secretions with areas of necrosis and calcification (Figures 3b and 3c). Genomic testing was not available in our hospital (or elsewhere in Ethiopia) for further confirmation of the histologic diagnosis or the fusion subtype.



Postoperative course
The patient was admitted to the ICU for the first 24 hours, then transferred to the ward for routine postoperative care. She developed a superficial surgical site infection on third post-operative day and culture showed coagulase-negative staphylococcus. She was started on a 7-day course of appropriate antibiotics and local wet to dry wound care. Chest tube was removed after 5 days and the patient discharged home on the 11th post operative day with healing of the surgical site. On follow-up outpatient visits, the patient is recovering well 6 months post operatively and has recently completed treatment with adjuvant chemotherapy.
DISCUSSION
SS, a rare mesenchymal tumor, accounts for 10% of all soft tissue sarcomas, and generally arises in the extremities.1,2 It is believed to originate from pluripotent mesenchymal tissue rather than the synovial cells because they occur in places where synovial structures do not exist and can show epithelial differentiation.3,4 Thus, the terminology of SS is a bit of a misnomer. Although we cannot be certain of the exact site of origin in our patient since the tumor involved the pleura, the mediastinum and upper retroperitoneum, it is probably most consistent with primary pleural SS (PPSS) which is an extremely rare tumor. The absence of extra-thoracic disease should be confirmed before the diagnosis of PPSS is made since SS, like other soft tissue sarcomas, tend to metastasize to the pleura and lung.4–7
The clinical symptoms of PPSS vary widely, mainly based on the histology and region of the tumor occurrence, as well as the size and degree of tumor differentiation.8 Initially, the patient may be asymptomatic but as the disease progresses, patients may present with non-specific pulmonary symptoms such as chest pain, cough, or dyspnea. In the current case, symptoms were predominantly abdominal due to the location of the tumor in the thoracoabdominal junction. The radiologic manifestations of PPSS overlap with many other neoplasms in the lung and pleura,9 including primary and metastatic lung cancer, solitary fibrous tumor of the pleura and other rare parenchymal sarcomas (e.g., leiomyosarcoma, sarcomatoid carcinoma, and malignant fibrous histiocytoma).9–11 In the present case, paraganglioma and primary esophageal tumor (i.e. leiomyoma or GIST) were the top diagnoses based on preoperative radiologic findings and endoscopy.
PPSS share the histologic, and immunohistochemical features of SS arising in other locations6,12 appearing as monophasic, biphasic, or poorly differentiated tumors. The two most common subtypes are monophasic spindle and biphasic.8 The unique fusion gene, SYT-SSX, generated by a specific chromosomal translocation t(x;18) (p11: q11) which fuses parts of chromosome 18 and the X chromosome. This has been detected in more than 95% of SS and currently represents the most specific evidence for definitive diagnosis. Additionally, this chromosomal translocation is now known to have two subtypes, commonly referred to SSX1 and SSX2. The SSX1 fusion is known to have a higher proliferation rate and poorer clinical outcomes.13 Fusion gene detection is increasingly being considered indispensable, with SS18 break-apart fluorescence in situ hybridization (FISH) being favored in many laboratories.14,15 Unfortunately, we do not have access to genomic testing at either our hospital or in Ethiopia.
PPSS appears to be more aggressive than SS arising in extremities with most patients dying within 1–3 years.4,11,16 Because of its rarity, the optimal treatment for PPSS remains unclear. Complete resection is considered the best option. Adjuvant chemotherapy and/or radiotherapy, and even neoadjuvant therapy have been used but again, due to the rarity of this tumor subset, definitive data are lacking.1,17 Repeat resections have been performed for local recurrence and sequenced resections are reported for cases in which the tumor could not be completely resected in a single operation.18 Fortunately, we were able to achieve a complete tumor resection in our patient but her follow-up is currently short and her prognosis uncertain. Longer follow-up with serial chest and abdominal CT imaging is essential.
CONCLUSION
PPSS is a rare, aggressive neoplasm that is difficult to confirm based on imaging, but should be considered in the differential diagnosis of pleural based thoracic tumors without extra thoracic sites of disease or regional lymphadenopathy. The benefit of adjuvant therapy following complete resection is uncertain but careful long-term follow-up is essential given the propensity of these tumors to recur.4,11,12,16
Conflict of interest
All authors declare no conflicts of interest
Informed consent
We obtained verbal and written informed consent with local language (Amharic) from the patient for surgical intervention as well as willingness to be reported for publication.